Simplified Molecular Input Line Entry System
Pour les articles homonymes, voir Smile.
Le Simplified Molecular Input Line Entry System ou SMILES est un langage symbolique de description des molécules chimiques sous forme de courtes chaînes de caractères ASCII. Les chaînes SMILES peuvent être importées dans la plupart des éditeurs de structure moléculaire pour être reconverties en représentation 2D ou en modèle 3D, et par conséquent, peuvent servir à constituer des bases de données.
La définition du langage SMILES a été introduite par David Weininger à la fin des années 1980[1]. Plusieurs extensions et modifications ont été ajoutées depuis, notamment par la société Daylight Chemical Information Systems, dont Weininger est le fondateur. Il existe d'autres langages à ligne de texte pour représenter les molécules, dont InChl, introduit par l'IUPAC. SMILES est cependant plus répandu, car plus facile à lire par l'utilisateur humain et surtout parce qu'il est implémenté dans de nombreux logiciels de chimie moléculaire.
Principes
[modifier | modifier le code]SMILES est bâti sur la représentation d'une molécule chimique sous forme d'un graphe, au sens mathématique du terme. On parle alors de graphe moléculaire. Les atomes sont les sommets du graphe, et les liaisons chimiques en sont les arêtes. Chaque sommet du graphe est étiqueté avec le symbole de l'atome correspondant (C, N, O…).
La chaîne de caractères SMILES correspondant à une molécule donnée est obtenue en parcourant le graphe suivant un algorithme de parcours en profondeur et en retenant le symbole de chaque sommet atteint. Le graphe de la molécule est préalablement simplifié en supprimant tous les atomes d'hydrogène (notation implicite) et en ouvrant les cycles pour transformer le graphe en arbre couvrant (spanning tree). À chaque fois qu'un cycle est ouvert, un suffixe numérique est ajouté pour indiquer la connexion des sommets correspondant à la liaison chimique supprimée. Des parenthèses sont utilisées pour indiquer les points de branchement sur l'arbre.
De par la nature de ce langage, il existe, pour une même molécule organique constituée de plusieurs atomes autres que l'hydrogène, plusieurs écritures possibles, selon l'atome de départ et l'ordre de parcours. Ainsi, CCO, OCC, C(C)O et C(O)C sont quatre représentations valides de la molécule d'éthanol CH3–CH2–OH.
Les principes de SMILES sont présentés dans le manuel du SMILES théorique[2] proposé par Daylight Chemical Information Systems[3].
Mise en œuvre et exemples
[modifier | modifier le code]
Atomes
[modifier | modifier le code]Les atomes sont représentés par leur symbole chimique entre crochets, comme [Se] pour le sélénium. Les crochets peuvent être omis pour les éléments usuels de la chimie organique : C, N, O, P, S, B, F, Cl, Br et I. Tous les autres éléments doivent être mis entre crochets. Lorsque les crochets sont omis, les valences libres de chaque atome sont supposées être complétées par des atomes d'hydrogène. Par exemple, la représentation SMILES de l'eau est O, celle de l'éthanol est CCO.
Un atome portant une ou plusieurs charges électriques est placé entre crochets, suivi du symbole H s'il est lié à un ou plusieurs atomes d'hydrogène (ceux-ci sont alors suivis de leur nombre [sauf s'il n'y en a qu'un] : NH4 pour l'ammonium), puis du signe '+' pour une charge positive, ou du signe '-' pour une charge négative. Le nombre des charges est alors spécifié après le signe (sauf s'il n'y en a qu'une) ; il est cependant aussi possible d'écrire le signe de la charge autant de fois que l'ion en possède : au lieu de "Ti+4", on peut très bien écrire "Ti++++" (Titane IV, Ti4+). Ainsi, l'anion hydroxyde est représenté par [OH-], le cation oxonium par [OH3+], et le cation cobalt III (Co3+) par [Co+3], ou [Co+++].
Liaisons
[modifier | modifier le code]La liaison simple est implicite et n'est pas représentée : CC représente donc l'éthane et CCC, le propane. La double liaison est représentée par le symbole "=" et la triple liaison par le symbole "#". L'éthylène est donc C=C, et le cyanure d'hydrogène, C#N.
Ramifications
[modifier | modifier le code]Les ramifications de la structure sont indiquées entre parenthèses, immédiatement après l'atome sur lequel elles sont branchées. Par exemple CC(=O)O représente l'acide acétique, avec deux atomes d'oxygène portés par le carbone du groupement carboxylique. Lorsque deux ramifications partent du même atome, la ramification suivante est placée immédiatement après la fin de parenthèse de la première : la formule du diméthylpropane sera donc CC(C)(C)C. Les ramifications peuvent être enchaînées et imbriquées pour décrire des structures moléculaires plus complexes, comme celle de l'acide citrique : OC(=O)CC(O)(CC(O)=O)C(O)=O.

Cycles
[modifier | modifier le code]La fermeture des cycles est indiquée par des chiffres placés après les atomes qui sont reliés. Le cyclohexane est représenté par C1CCCCC1, les deux "1" servant de labels indiquant que les deux carbones qui les précèdent sont reliés, formant un cycle à six atomes. De même, la formule SMILE du 1,1-diméthylcyclopentane sera C1CCCC(C)(C)1 ; celle du 3-cyanoanisole (3-methoxybenzonitrile) pourra être COc(c1)cccc1C#N (voir ci-contre). Pour un deuxième cycle, le label sera 2 (naphtalène : c1cccc2c1cccc2) ; au-delà de 9, le signe '%' doit précéder le label, afin de le différencier de deux labels distincts liés au même atome (~C12~ signifiera que l'atome de carbone porte les liaisons de fermeture de cycle 1 et 2, tandis que ~C%12~ indiquera un seul label, le 12).
Aromaticité
[modifier | modifier le code]Les atomes C, N, O, S participant à un cycle aromatique sont représentés en minuscule par 'c', 'n', 'o' et 's', respectivement. Les doubles liaisons sont rarement indiquées : le benzène est ainsi représenté par c1ccccc1 (plutôt que par C1=CC=CC=C1, qui correspond à la formule de Kekulé), et le furane c1cocc1.
Les liaisons entre des atomes aromatiques sont considérées par défaut comme aromatiques, bien que cette liaison aromatique puisse être explicitement spécifiée avec le symbole ':'. De fait, pour indiquer une liaison simple entre deux anneaux (ou atomes) aromatiques, on doit la représenter par son symbole '-', habituellement omis afin de ne pas surcharger la séquence : le biphényle sera donc représenté par c1ccccc1-c2ccccc2.
Un atome d'azote aromatique lié à un atome d'hydrogène, tel celui de la molécule de pyrrole, doit être représenté [nH] ; le pyrrole sera donc représenté c1c[nH]cc1, et l'imidazole n1c[nH]cc1.
Les algorithmes de Daylight et de OpenEye, permettant de générer des chaînes SMILES canoniques différant dans leur traitement de l'aromaticité.
Il convient de noter que l'aromaticité telle qu'elle est détectée par les différents interpréteurs de chaînes SMILES est souvent très différente de l'aromaticité réelle des composés.
Stéréochimie
[modifier | modifier le code]
La configuration des doubles liaisons carbone-carbone est représentée par des caractères '/' et '\', placés autour des carbones concernés. Ainsi, F/C=C/F est une représentation du trans-1,2-difluoroéthène (les atomes de fluor ne se trouvent pas « en face » l'un de l'autre), et F/C=C\F une représentation du cis-1,2-difluoroéthène (les atomes de fluor se trouvent du même côté par rapport à l'axe de la double liaison). Cependant, le fait que le deuxième slash ait la même orientation '/' ou non '\' ne signifie pas que la double liaison est trans ou cis, mais simplement que le premier radical suivant la double liaison est du même côté que celui qui la précède (slashs opposés « /C=C\ » ou « \C=C/ ») ou du côté opposé (slashs de même sens). Ainsi, le trans-2-hydroxybut-2-ène aura pour formule possible C/C=C(/C)O, ou encore C/C=C(\O)C, et le cis-2-hydroxybut-2-ène deviendra C/C=C(\C)O.
La configuration des atomes de carbones asymétriques et des centres chiraux est, quant à elle, indiquée par le symbole '@'. L'exemple le plus commun est la L-Alanine (acide 2S-aminopropanoïque), énantiomère représenté par la formule N[C@@H](C)C(=O)O. Ici, « @@ » signifie que les radicaux –H, –CH3 et –C(=O)OH sont disposés dans le sens des aiguilles d'une montre autour du carbone chiral, si l'on observait celui-ci depuis la position de l'atome d'azote. les radicaux –H n'étant pas directement représentés dans la notation SMILES, si un carbone chiral en possède un, il doit être accolé aux arobases, entre les crochets « [] », et se trouve donc en première place dans l'ordre des radicaux. Écrire N[C@@H](C)C(=O)O revient donc à écrire N[C@@][H](C)C(=O)O. À l'inverse, un seul symbole '@' indique une lecture dans le sens inverse des aiguilles d'une montre (sens antihoraire) des radicaux suivants l'atome chiral. La D-Alanine (acide 2R-aminopropanoïque), reflet de son homologue L vue à travers un miroir, peut donc être écrite N[C@H](C)C(=O)O ou encore N[C@@H](C(=O)O)C (le fait d'échanger l'ordre des radicaux méthyle et carboxyle a le même effet qu'inverser le sens de rotation).
Isotopes
[modifier | modifier le code]Un isotope peut être spécifié en plaçant l'atome concerné entre crochets et en faisant précéder son symbole par son nombre de masse. Ainsi, une molécule de benzène dotée d'un atome de carbone-14 pourra être écrite [14C]1ccccc1, tandis que le chloroforme deutérié sera [2H]C(Cl)(Cl)Cl.
Autres possibilités
[modifier | modifier le code]Deux molécules ou ions distincts peuvent être représentés dans une même formule SMILES. Leurs formules sont alors séparées par le caractère '.'. Ainsi, le chlorure de sodium ne sera pas représenté par [Na]Cl, ni par [Na+][Cl-] mais par [Na+].[Cl-] (il s'agit d'une liaison ionique).
Par ailleurs, il est possible de représenter des équations chimiques, à l'aide du caractère '>', réactifs >> produits pour une réaction simple et réactifs > agent > produits pour une réaction faisant intervenir un agent qui n'y participera pas directement (catalyseur). C=C.O=O.O=O.O=O>>O=C=O.O=C=O.O sera donc équivalent à CH2=CH2 + 3 O2 → 2 CO2 + H2O (combustion de l'éthène).
Application à diverses structures
[modifier | modifier le code]| Molécule | Structure | Formule SMILES |
|---|---|---|
| Diazote | N≡N | N#N |
| Isocyanate de méthyle (MIC) | CH3–N=C=O | CN=C=O |
| Sulfate de cuivre | Cu2+ SO42− | [Cu+2].[O-]S(=O)(=O)[O-] |
| Énanthotoxine (C17H22O2) | 
|
CCC[C@@H](O)CC\C=C\C=C\C#CC#C\C=C\CO |
| Pyréthrine II (C21H28O5) | 
|
COC(=O)C(\C)=C\C1C(C)(C)[C@H]1C(=O)O[C@@H]2C(C)=C(C(=O)C2)CC=CC=C |
| Aflatoxine B1 (C17H12O6) | 
|
O1C=C[C@H]([C@H]1O2)c3c2cc(OC)c4c3OC(=O)C5=C4CCC(=O)5 |
| Glucose (glucopyranose) (C6H12O6) | 
|
OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)1 |
| Cuscutine alias Bergenin (résine) (C14H16O9) | 
|
OC[C@@H](O1)[C@@H](O)[C@H](O)[C@@H]2[C@@H]1c3c(O)c(OC)c(O)cc3C(=O)O2 |
| Une phéromone de la cochenille californienne | 
|
CC(=O)OCCC(/C)=C\C[C@H](C(C)=C)CCC=C |
| 2S,5R-Chalcogran : phéromone du scolyte Pityogenes chalcographus[4] | ![(2S, 5R)-2-ethyl-1,6-dioxaspiro[4.4]nonane](http://upload.wikimedia.org/wikipedia/commons/thumb/8/8e/2S%2C5R-chalcogran-skeletal.svg/260px-2S%2C5R-chalcogran-skeletal.svg.png)
|
CC[C@H](O1)CC[C@@]12CCCO2 |
| Vanilline | 
|
O=Cc1ccc(O)c(OC)c1 |
| Mélatonine (C13H16N2O2) | 
|
CC(=O)NCCC1=CNc2c1cc(OC)cc2 |
| Flavopéréirine (C17H15N2) | 
|
CCc(c1)ccc2[n+]1ccc3c2Nc4c3cccc4 |
| Nicotine (C10H14N2) | 
|
CN1CCC[C@H]1c2cccnc2 |
| Alpha-thujone (C10H16O) | 
|
CC(C)[C@@]12C[C@@H]1[C@@H](C)C(=O)C2 |
| Thiamine (C12H17N4OS+) (vitamine B1) |
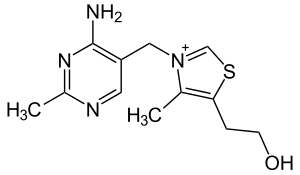
|
OCCc1c(C)[n+](cs1)Cc2cnc(C)nc(N)2 |
Illustration avec une molécule de plus de neuf cycles, la Céphalostatine-1[5] (molécule stéroïdique de formule brute C54H74N2O10 issue d'un ver marin de la famille des Hydrophiloidea, Cephalodiscus gilchristi) :

Donnera, en partant du radical méthyle le plus à gauche sur la figure :
C[C@@](C)(O1)C[C@@H](O)[C@@]1(O2)[C@@H](C)[C@@H]3CC=C4[C@]3(C2)C(=O)C[C@H]5[C@H]4CC[C@@H](C6)[C@]5(C)Cc(n7)c6nc(C[C@@]89(C))c7C[C@@H]8CC[C@@H]%10[C@@H]9C[C@@H](O)[C@@]%11(C)C%10=C[C@H](O%12)[C@]%11(O)[C@H](C)[C@]%12(O%13)[C@H](O)C[C@@]%13(C)CO
(Remarquez les '%' devant l'indice des labels de fermeture de cycle supérieur à 9, voir la section « Cycles », plus haut).
Conversion
[modifier | modifier le code]Il existe des algorithmes de transformation du schéma d'une molécule en chaîne SMILES. Comme il n'existe en général pas de description unique d'une molécule, il y a également plusieurs SMILES pouvant décrire la même structure : tout dépend de l'atome dont on part, de l'ordre dans lequel on décrit les branches du graphe moléculaire et de la manière dont on ouvre les cycles. Par exemple, c1ccccc1o et c1ccc(o)cc1 sont deux représentations équivalentes du phénol.
Réciproquement, la description SMILES d'une molécule peut être reconvertie en représentation chimique 2D classique, au moyen d'algorithmes de représentation de graphe.
La méthode SMILES permet d'échanger numériquement des descriptions de molécules arbitrairement complexes par le biais d'un fichier texte classique. La représentation SMILES étant très compacte, elle permet de stocker simplement des fichiers de plusieurs centaines de milliers de molécules (chimiothèques).
Extensions
[modifier | modifier le code]SMILES Isomérique
[modifier | modifier le code]Des extensions du langage SMILES permettent de décrire la chiralité et la conformation des doubles liaisons. On peut ainsi décrire la conformation R ou S d'un carbone asymétrique ou l'isomérie cis/trans. On peut également décrire les états de protonation, la présence d'isotopes spécifiques et même décrire des schémas réactionnels.
SMARTS
[modifier | modifier le code]SMARTS est une extension qui permet, en plus des définitions classiques, de remplacer des atomes ou des liaisons par des jokers. Ceci est utilisé pour spécifier des motifs chimiques utilisés pour des recherches dans des banques de données de molécules (chimiothèques). Cela permet en particulier la recherche de sous-structures chimiques communes à deux molécules.
Voir aussi
[modifier | modifier le code]Références
[modifier | modifier le code]- D. Weininger ; SMILES, « a Chemical Language and Information System. 1. Introduction to Methodology and Encoding Rules » (1988) J. Chem. Inf. Comput. Sci. 28:31-36
- Manuel du SMILES théorique.
- Daylight Chemical Information Systems.
- [PDF] Isolation of pheromone synergists of bark beetle, Pityogenes chalcographus, from complex insect-plant odors by fractionation and subtractive-combination bioassay
- PubChem Compound CID=183413 (Céphalostatine-1)