Амины
| Первичный амин | Вторичный амин | Третичный амин |
|---|---|---|
 |
 |
 |
Ами́ны — органические соединения, являющиеся производными аммиака, в молекуле которого один или несколько атомов водорода замещены на углеводородные радикалы. По числу замещённых атомов водорода различают соответственно первичные (замещён один атом водорода), вторичные (замещены два атома из трёх) и третичные амины (замещены все три атома). Выделяют также четвертичные аммониевые соединения вида R4N+X-[1].
По характеру органической группы, связанной с азотом, различают алифатические, ароматические и жирно-ароматические (содержат ароматический и алифатический радикалы) амины. Ароматические амины называют анилинами. По числу NH2-групп в молекуле амины делят на моноамины, диамины, триамины либо полиамины[1].
Номенклатура
[править | править код]Рекомендации ИЮПАК предписывают следующие правила для составления названий аминов. В случае первичных аминов пользуются одним из трёх способов: (1) добавляют суффикс «-амин» к названию родоначального углеводорода (предпочтительно); (2) добавляют название заместителя к корню «азан» или (3) добавляют название заместителя к корню «амин». Например:
- (1) CH3NH2 — метанамин;
- (2) CH3NH2 — метилазан;
- (3) CH3NH2 — метиламин[2].
Для вторичных и третичных аминов используются похожие рекомендации: (1) составить заместительное название с суффиксом «-амин» и указать остальные заместители при атоме азота (предпочтительно); (2) указать заместители в алфавитном порядке в виде приставок к корню «азан» или (3) указать заместители в алфавитном порядке в виде приставок к корню «амин». Например:
- (1) (CH3CH2)2NCH2CH3 — N,N-диэтилэтанамин;
- (2) (CH3CH2)2NCH2CH3 — триэтилазан;
- (3) (CH3CH2)2NCH2CH3 — триэтиламин[2].
В более сложных структурах, где аминогруппа не является старшей, она обозначается в виде префикса «амино-» (H2NCH2CH2COOH — 3-аминопропановая кислота). Если эта аминогруппа дополнительно замещена, название заместителя помещают перед ней в виде приставки ((CH3NH)2CHCH2CH2COOH — 4,4-бис(метиламино)бутановая кислота)[2].
Диамины, триамины и т. д. называют, добавляя перед суффиксом «-амин» множащие приставки «ди-», «три-», «тетра-» и т. д. (H2NCH2CH2NH2 — этандиамин-1,2, этилендиамин)[2].
Многие ароматические амины сохраняют тривиальные названия: анилин PhNH2, толуидины CH3C6H4NH2, анизидины CH3OC6H4NH2[3].
Физические свойства и строение
[править | править код]Физические свойства аминов
[править | править код]Низшие амины — метиламин, диметиламин, триметиламин и этиламин — при комнатной температуре являются газами. Высшие амины до 12 атомов углерода являются жидкостями. Амины с более длинными заместителями являются твёрдыми веществами[4].
Низшие амины смешиваются с водой. Трибутиламин смешивается с водой частично[4].
Амины имеют характерный рыбный запах, который можно почувствовать при концентрации амина 0,1 м. д.[4]
Строение аминогруппы
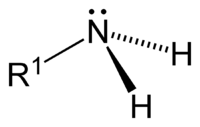
[править | править код]Аминогруппа имеет пирамидальное строение: пирамиду образуют три заместителя атома азота, а в четвёртой вершине тетраэдра находится неподелённая электронная пара. Длина связи N–H в метиламине равна 1,011 Å, а длина связи C–N составляет 1,474 Å. Угол H–N–H равен 105,9°, а угол C–N–H равен 112,9°[5].
Инверсия атома азота
[править | править код]
Имея тетраэдрическое строение, sp3-гибридный атом азота в аминах постоянно претерпевает инверсию через sp2-гибридное состояние. Энергетический барьер для инверсии у алкиламинов составляет 16-40 кДж/моль. При комнатной температуре скорость инверсии оставляет 103−105 Гц. Это приводит к тому, что если амин имеет три разных заместителя при атоме азота, теоретически для него можно изобразить энантиомерные структуры, однако на практике их выделить нельзя, потому что из-за инверсии они быстро превращаются друг в друга. Исключением является основание Трёгера, в котором конфигурация атомов азота закреплена и которое существует в виде двух стереоизомеров[6]. Также стереохимически стабильны четвертичные аммониевые соли[5].
Спектральные характеристики
[править | править код]ИК-спектры аминов характеризуются наличием полос, соответствующих колебаниями связей N-H. У первичных аминов эти колебания проявляются в виде двух полос в области 3400-3380 см−1 и 3340-3320 см−1 (полосы соответствуют симметричным и антисимметричным колебаниям N-H). У вторичных аминов есть только одна полоса в области 3360-3310 см−1. Третичные амины не имеют полос поглощения в этой области. Ароматические амины имеют соответственно то же число полос в области 3500-3300 см−1[3].
Алифатические амины не поглощают в видимой и ультрафиолетовой области спектра. Ароматические амины имеют две полосы поглощения, соответствующие π→π*-переходам[3].
Получение
[править | править код]Из спиртов
[править | править код]Стандартным промышленным способом получения низших аминов является реакция соответствующего спирта с аммиаком над подходящим катализатором. Поскольку получаемый первичный амин может также реагировать со спиртом, продуктом всегда является смесь первичного, вторичного и третичного амина. Кроме того, образование вторичного и третичного амина является экзотермическим, а поэтому выгодным. Состав продуктов можно контролировать соотношением реагентов, температурой и продолжительностью синтеза[7].
Аммиак, спирт и водород пропускают над катализаторами на основе никеля, кобальта, меди, железа, реже — платины и палладия. В качестве подложки применяются оксид алюминия, оксид кремния и оксид циркония. Условия проведения реакции: 0,5–25 МПа, 100–250 °С (в зависимости от катализатора). Считается, что процесс протекает в три стадии:
- дегидрирование спирта до альдегида;
- реакция карбонильного соединения и амина с образованием имина;
- гидрирование имина до амина[7].
Чтобы сдвинуть равновесие в сторону первичных аминов, используют двойной избыток аммиака. Согласно уравнению реакции, дополнительный водород в ней не требуется, однако в его отсутствие происходит образование побочных продуктов: иминов, енаминов и нитрилов. Наличие водорода также способствует активности катализатора[7].
Из карбонильных соединений
[править | править код]По аналогии с предыдущим способом, амины получают по реакции аммиака с карбонильными соединениями. В этом случае продукт необходимо прогидрировать, поэтому водород расходуется в стехиометрическом количестве. Катализаторы используются те же, что и в синтезе аминов из спиртов[8].
Из нитрилов
[править | править код]Нитрилы в промышленности каталитически гидрируют до соответствующих первичных аминов. В качестве катализаторов используют благородные металлы (палладий, платину, родий), никель, кобальт, а также железо. Благородные металлы позволяют провести реакцию в мягких условиях: 20—100 °С, 0,1–0,5 МПа, а никелевые и кобальтовые катализаторы требуют температуры до 180 °С и давления в 25 МПа[8].
Из Галогенпроизводных
[править | править код]По реакции Делепина (через гексамин)


Другие промышленные методы
[править | править код]Традиционный препаративный подход, основанный на реакции алкилгалогенидов и аммиака или аминов с образованием аммониевых солей, не нашёл широкого применения в промышленности. В настоящее время так получают лишь этилендиамин, гомологичные ему полиамины, аллиламин и некоторые малотоннажные лекарственные препараты. Проблемой в данном подходе является отсутствие дешёвого сырья, коррозия, а также проблемы с качеством продуктов[9].
Восстановление нитросоединений используется редко, поскольку исходные нитроалканы не очень широко доступны. По состоянию на 2015 год этот метод применяется для синтеза 2-амино-2-метилпропанола-1[9]. Первичные ароматические амины получаются по этому методу хорошо: наиболее часто для их получения ароматические нитросоединения гидрируют водородом в жидкой или газовой фазе в присутствии никеля, платины или палладия. Также применяют железо или цинк и сульфиды щелочных металлов[3].
Амины с третичным углеводородным заместителем, например, трет-бутиламин весьма трудно получить обычными методами. Их синтезируют по реакции Риттера, присоединяя циановодород к алкену в присутствии концентрированной серной кислоты. Процесс проводят при 30—60 °С, а получаемый полупродукт гидролизуют при 100 °С. Применение реакции Риттера весьма ограниченно из-за использования токсичного циановодорода, а также из-за образования существенного количества побочных солей (3,3 кг на 1 кг трет-бутиламина), которые необходимо утилизировать[9].
Лабораторные методы
[править | править код]В лабораторных условиях амины получают разнообразными методами: синтезом по Габриэлю, восстановлением нитрилов водородом, алюмогидридом лития либо дибораном, восстановлением амидов под действием тех же реагентов, восстановлением азидов, оксимов и нитросоединений[10].
Первичные и вторичные амины удобно получать по реакции восстановительного аминирования. Для синтеза первичных аминов в реакцию вводят карбонильное соединение и аммиак, а для синтеза вторичных аминов — карбонильное соединение и первичный амин. Полученное основание Шиффа затем восстанавливают водородом, боргидридом натрия или цианоборгидридом натрия[10].
Кроме того, первичные амины можно получить из карбоновых кислот при помощи перегруппировок Гофмана, Шмидта и Курциуса[10].
Одним из лабораторных способов является реакция аминов и аммиака с галогеналканами:
Такие реакции, которые наиболее полезны для алкилйодидов и бромидов, редко используются, поскольку степень алкилирования трудно контролировать[11]. Селективность может быть улучшена с помощью реакции Делепина, хотя это редко используется в промышленном масштабе.
Прямое электрофильное аминирование
[править | править код]Получение ароматических аминов прямым электрофильным аминированием ароматических углеводородов долгое время считалась неосуществимым. В 2019 году российские ученые из Томского политехнического университета показали возможность прямого аминирования аренов гидразойной кислотой по классическому механизму SEAr, с участием катиона аминодиазония H2N3+[12].
Химические свойства
[править | править код]Основные свойства
[править | править код]Амины, являясь производными аммиака, имеют сходное с ним строение и проявляют подобные ему свойства. Атом азота содержит неподелённую электронную пару и выступает как основание Льюиса. Амины являются более сильными основаниями, чем вода, поэтому они также проявляют свойства оснований Брёнстеда — Лоури. Численно основные свойства аминов выражаются константой основности Kb либо pKb[13].
Амины являются более сильными основаниями, чем аммиак, за счёт донорного влияния алкильных групп. Однако из всех аминов наиболее сильными основаниями являются вторичные амины. Третичные амины проигрывают им в основности, что связано с пространственными препятствиями для переноса к ним протона и последующей сольватации образовавшегося аммониевого катиона. В газовой фазе, где эффекты сольватации отсутствуют, основность аминов предсказуемо уменьшается в следующем ряду: третичные > вторичные > первичные > аммиак.[13].
Однако в водных растворах эта закономерность искажается, наличие третьего заместителя создаёт пространственное затруднение как для присоединения протона, так и для сольватации образовавшегося катиона молекулами растворителя.
Ароматические амины являются более слабыми основаниями, что связывают с делокализацией неподелённой пары атома азота по ароматическому ядру[13].
| название | формула | pKb | pKa(BH+) | pKa |
|---|---|---|---|---|
| диэтиламин | Et2NH | 3.06 | 10.94 | |
| триэтиламин | Et3N | 3.25 | 10.75 | |
| диметиламин | (CH3)2NH | 3.27 | 10.73 | |
| метиламин | (CH3)NH2 | 3.36 | 10.64 | |
| этиламин | EtNH2 | 3.37 | 10.63 | |
| триметиламин | (CH3)3N | 4.19 | 9.81 | |
| аммиак | NH3 | 4.79 | 9.21[14] | ~33 |
| 4-метоксиланилин | 8.66 | 5.34 | ||
| 4-метиланилин | 8.90 | 5.10 | 8.83 | |
| анилин | 9.38 | 4.62 | ||
| 4-хлоранилин | 10.02 | 3.98 | ||
| 4-нитроанилин | 4-NO2C5H4NH2 | 13 | 1.0 |
| соединение | Алкиламины | NH3 | Ариламины |
|---|---|---|---|
| pKa(BH+) | 10.6 - 11.2 | 9.26 | 4.6 - 5.1 |
Амины являются очень слабыми кислотами: pKa для них составляет порядка 35-40. Соответственно, анионы, получаемые из аминов, являются очень сильными основаниями, что находит применение в органическом синтезе (см. LDA)[13].
Алкилирование аминов
[править | править код]Амины реагируют с алкилгалогенидами по механизму нуклеофильного замещения с образованием более замещённых аминов. Реакция протекает в диполярных апротонных растворителях (ДМФА, ацетонитриле)[15].
Ацилирование аминов
[править | править код]Первичные и вторичные амины вступают в реакции ацилирования с галогенангидридами, ангидридами карбоновых кислот, сложными эфирами. Ацилирующие реагенты можно расположить в ряд активности: RCOR < RCONR2 < RCOOR < (RCO)2O < RCOHal < RCOBF4. Также скорость реакции зависит от нуклеофильности амина, которую условно можно связать с основностью амина: алкиламины > ариламины > амиды. Внутримолекулярное ацилирование происходит легче, чем межмолекулярное[16].

В реакции с хлорангидридами происходит выделение хлороводорода, поэтому в реакцию необходимо брать двойное количество амина, чтобы второй эквивалент связал этот хлороводород. Образующаяся аммониевая соль выпадает в осадок и фильтруется. Как следствие, максимальный выход амида из амина составляет 50 %. Как вариант, можно использовать другие органические и неорганические основания, чтобы повысить выход. Например, в реакции Шоттена — Баумана используется гидроксид натрия или гидроксид калия. Из органических оснований применяются пиридин, диметиланилин, триэтиламин и др.[16]
Реакция с альдегидами и кетонами
[править | править код]При реакции с альдегидами и кетонами первичные амины образуют имины (основания Шиффа), а вторичные амины дают енамины[15].
Реакция с азотистой кислотой
[править | править код]Реакция с азотистой кислотой является качественной для идентификации первичных, вторичных и третичных аминов. Первичные алифатические амины диазотируются азотистой кислотой с образованием солей алкилдиазония. Эти соли даже в мягких условиях разлагаются с выделением газообразного азота и образованием карбокатиона, который может превратиться в алкен, спирт или другое устойчивое соединение[15]. Иногда эту реакцию используют для расширения цикла, как, например, в перегруппировке Демьянова[3].
В отличие от алифатических, первичные ароматические амины при диазотировании образуют устойчивые соли арендиазония, которые способны вступать в реакции замещения молекулы азота на нуклеофил либо в реакции азосочетания[15].
Вторичные амины (алифатические и ароматические) при действии азотистой кислоты нитрозируются по атому азота, давая жёлтые N-нитрозоамины[15][3].
Алифатические третичные амины дают смесь соли амина и N-нитрозоаммонийной соли[15], а ароматические третичные амины нитрозируются в пара-положение[3].
Реакция с сульфонилгалогенидами
[править | править код]Реакция аминов с бензолсульфонилхлоридом либо пара-толуолсульфонилхлоридом также является качественной реакцией на первичные, вторичные и третичные амины и называется тестом Хинсберга. В этой реакции смешивают амин и сульфогалогенид и встряхивают их с водным раствором гидроксида натрия, а через 10-15 мин подкисляют полученный раствор. Первичные амины на первой стадии дают сульфамид RNHSO2Ar, который в щёлочи растворяется благодаря наличию кислого атома водорода при атоме азота. При добавлении кислоты он выпадает в осадок[15].
Вторичные амины также дают сульфамид, однако он не содержит кислого атома водорода и не растворяется в щёлочи. При подкислении смеси в данном случае ничего не происходит[15].
Третичные амины не вступают в эту реакцию, а сульфонилгалогенид в щелочной среде гидролизуется до соли сульфокислоты. При подкислении третичный амин растворяется, переходя в солевую форму[15].
Галогенирование аминов
[править | править код]Под действием гипохлоритов первичные и вторичные амины галогенируются по атому азота[15].
Окисление аминов
[править | править код]Амины легко вступают в реакции окисления, причём легче всего это делают третичные амины. Препаративными реагентами для этого превращения являются раствор пероксида водорода и органические надкислоты. Образующиеся N-оксиды третичных аминов бывают хиральными и могут быть разделены на энантиомеры. При обработке восстановителями, например трифенилфосфином, они снова превращаются в амины[15].
Первичные амины могут окисляться до нитросоединений. В качестве окислителя в этой реакци используют трифторнадуксусную кислоту[15].
Реакции ароматических аминов
[править | править код]Ароматические амины (анилины) вступают в типичные реакции ароматического электрофильного замещения. Поскольку аминогруппа является активирующим заместителем, эти реакции протекают очень активно даже под действием мягких реагентов[15].
Так, галогенирование анилинов не требует использования кислоты Льюиса. Реакцию нельзя остановить на стадии моно- и дигалогенирования: например, при бромировании образуется сразу 2,4,6-триброманилин. Если необходимо ввести только один атом галогена, аминогруппу ацилируют, уменьшая её активирующее влияние[15].
В стандартных условиях нитрования ароматических соединений амины быстро окисляются, поэтому нитруют их ацильные производные. Третичные амины можно нитровать азотной кислотой в уксусной кислоте. Сульфируют анилины "методом запекания": сначала смешивают амин с серной кислотой, получая соль, которую далее в сухом виде нагревают при 180-200 °С. В промышленности так получают сульфаниловую кислоту[15].
Защитные группы для аминов
[править | править код]В органическом синтезе применяется большое разнообразие защитных групп для аминов. Наибольшую популярность получили группы Cbz (бензилоксикарбонильная) и Boc (трет-бутоксикарбонильная). Бензилоксикарбонильную группу вводят обработкой амина бензиловым эфиром хлоругольной кислоты PhCH2OCOCl в присутствии основания. Удалить её можно гидрогенолизом либо бромоводородом в уксусной кислоте. Группа Boc вводится с помощью ди-трет-бутилдикарбоната, а удаляется при обработке кислотой[17].
Методы определения
[править | править код]Для идентификации аминов используют несколько качественных реакций. Первичные амины нагревают с хлороформом в присутствии щёлочи: при этом они превращаются в изонитрилы и дают неприятный запах. Вторичные амины обрабатывают азотистой кислотой, а полученный осадок сплавляют с фенолом и подкисляют, наблюдая зелёное окрашивание[18].
Количественное определение проводят методом Кьельдаля, методом Ван Слайка, бромометрией, кислотно-основным титрованием и хроматографией. Первичные амины также превращают в азосоединения или основания Шиффа, а затем анализируют фотометрически[18].
Вредное воздействие
[править | править код]Алифатические амины оказывают негативное действие на нервную систему и сосуды, нарушают проницаемость клеточных мембран, работу печени и вызывают развитие дистрофии. Ароматические амины способствуют выработке метгемоглобина; некоторые из них канцерогенны[18].
Примечания
[править | править код]- ↑ 1 2 Ullmann, 2015, p. 1–2.
- ↑ 1 2 3 4 Favre H. A., Powell W. H. Nomenclature of Organic Chemistry. IUPAC Recommendations and Preferred Names 2013. — The Royal Society of Chemistry, 2014. — P. 669–676. — doi:10.1039/9781849733069-FP001.
- ↑ 1 2 3 4 5 6 7 Химическая энциклопедия, 1988, с. 147.
- ↑ 1 2 3 Lawrence, 2004, p. 33–34.
- ↑ 1 2 Реутов, 2010, с. 276–278.
- ↑ Lawrence, 2004, p. 27–28.
- ↑ 1 2 3 Ullmann, 2015, p. 6–7.
- ↑ 1 2 Ullmann, 2015, p. 8.
- ↑ 1 2 3 Ullmann, 2015, p. 9.
- ↑ 1 2 3 Реутов, 2010, с. 287–299.
- ↑ Fritz Ullmann. Amines, Aliphatic // Ullmann's Encyclopedia of Industrial Chemistry. — 2000. — ISBN 3527306730.
- ↑ Ksenia S. Stankevich, Alexander A. Bondarev, Anastasia K. Lavrinenko, Victor D. Filimonov. Mechanism of Direct Electrophilic Aromatic Amination: an Electrophile is Found by Quantum-Chemical Study (англ.) // ChemistrySelect. — 2019. — Vol. 4, iss. 10. — P. 2933–2940. — ISSN 2365-6549. — doi:10.1002/slct.201803911. Архивировано 25 мая 2019 года.
- ↑ 1 2 3 4 Реутов, 2010, с. 282–287.
- ↑ Hall, H. K. (1957). Correlation of the Base Strengths of Amines1. Journal of the American Chemical Society, 79(20), 5441–5444. doi:10.1021/ja01577a030
- ↑ 1 2 3 4 5 6 7 8 9 10 11 12 13 14 15 Реутов, 2010, с. 300–321.
- ↑ 1 2 The Chemistry of Amides : [англ.] / Ed. Jacob Zabitsky. — Interscience Publishers, 1970. — P. 74–81. — ISBN 0471980498.
- ↑ Реутов, 2010, с. 322–323.
- ↑ 1 2 3 Химическая энциклопедия, 1988, с. 149.
Литература
[править | править код]- Салов Б. В. Амины // Химическая энциклопедия : в 5 т. / Гл. ред. И. Л. Кнунянц. — М.: Советская энциклопедия, 1988. — Т. 1: А — Дарзана. — С. 147–149. — 623 с. — 100 000 экз. — ISBN 5-85270-008-8.
- Реутов О. А., Курц А. Л., Бутин К. П. Органическая химия : в 4 т.. — 2-е изд. — М. : БИНОМ. Лаборатория знаний, 2010. — Т. 3, 19. Амины. — С. 273–324. — 2000 экз. — ISBN 978-5-9963-1335-8.
- Roose P., Eller K., Henkes E., Rossbacher R., Höke H. Amines, Aliphatic (англ.) // Ullmann's Encyclopedia of Industrial Chemistry. — Wiley, 2015. — doi:10.1002/14356007.a02_001.pub2.
- Lawrence S. A. Amines. Synthesis, Properties and Applications. — Cambridge University Press, 2004. — ISBN 0521782848.
Ссылки
[править | править код]- Амины // Энциклопедический словарь Брокгауза и Ефрона : в 86 т. (82 т. и 4 доп.). — СПб., 1890—1907.
- Физические свойства аминов